

https://doi.org/10.1038/s42005-022-00940-0
“Time-resolved studies with temporal resolution that separate molecular level dynamics from macroscopic changes, allow clear distinction between the time scales of the different degrees of freedom involved. Cooperative molecular switching in the solid state is exemplified by spin crossover phenomenon in crystals of transition metal complexes. Here we show the existence of a delay between the crystalline volume increase, and the cooperative macroscopic switching of molecular state. Using 100 ps X-ray diffraction, we track the molecular spin state and the structure of the lattice during the photoinduced low spin to high spin transition in microcrystals of [FeIII(3-MeO-SalEen)2]PF6. Model simulations explain the phenomenon with thermally activated kinetics governed by local energy barriers separating the molecular states. Such behaviour is different from that encountered in materials with no local energy barriers, where phase transformation can occur simultaneously with propagation of strain. Broadly, this motivates an optimised material design, scalable with size and intrinsic energetics.”
”
Phase transition and volume change in bulk single crystal
In this study, we focus on an SCO compound [FeIII(3-MeO-SalEen)2]PF6. The SCO materials serve as prototypical examples of cooperative switching between two molecular electronic states, Low Spin (LS) and High Spin (HS). The sample was previously identified as suitable candidate for the photo-induced out-of-equilibrium studies in solid-state from the angle of elastic properties that lead to strong cooperativity18. The crystalline structure of this material was characterized in previous studies (Supplementary Note 120,21). At the molecular level, the switching of electronic state of FeIII system, from LS (S = 1/2) to HS (S = 5/2) causes an increase of molecular volume, due to elongation of the Fe-Ligand bonds by around 0.15 Å between LS and HS states. The molecules in this compound are arranged in a closed-packed network20 (Fig. 1a). It is known that the relative stability between different macroscopic phases in such SCO crystals is ensured by the elastic intermolecular interactions of various strengths, resulting in more or less cooperative transformations22,23. In the present case, a strongly first-order phase transition is observed around 165 K, with a thermal hysteresis of 3 K, between the low-temperature LS phase and the high-temperature HS phase20. This phase transition is isostructural, as it does not imply any change of symmetry (same space group P-1 and Z = 2 for each phase), in a way similar to the gas-liquid transition24. The volume is a totally symmetric parameter and plays a central role for an isostructural phase transition. At thermal equilibrium, both the unit cell volume and the concentration of HS molecules show correlated jumps at the phase transition. This is usually observed for two totally symmetric degrees of freedom involved in changes associated with a phase transition without symmetry change25.
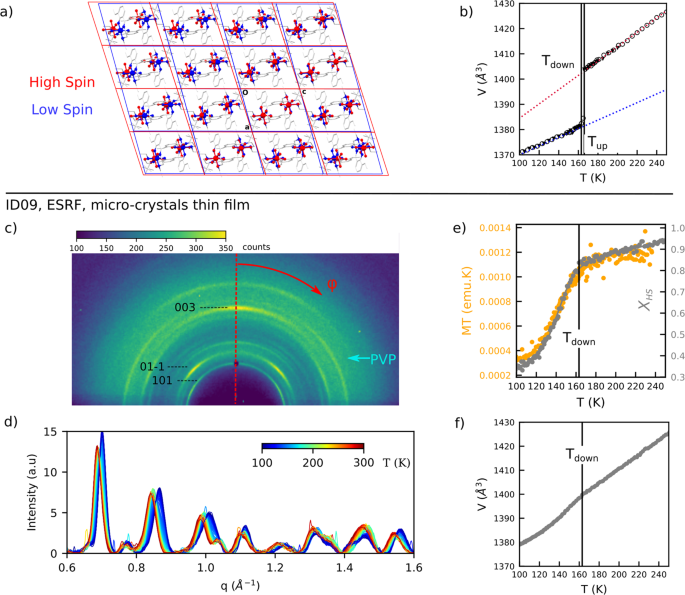
a [FeIII(3-MeO-SalEen)2]PF6 molecular lattices in Low Spin, LS (100 K, blue) and High Spin, HS (293 K, red) states, shown in the (a, c) plane. Ligands (in grey) are shown for the LS structure only. b X-ray diffraction (XRD) measurement on a single crystal: evolution on temperature, T, of the unit cell volume, V (T decreasing, plain black triangles, and T increasing, open circles) for extracting the thermal expansion parameters of the HS (red dotted line) and LS (blue dotted line) unit cell (Supplementary Note 120). Vertical black lines represent transition temperatures for T decreasing (Tdown) and T increasing (Tup). c–f Synchrotron x-ray powder diffraction on micro-crystals measured at ESRF, ID09 beamline: c Diffraction image measured at room temperature. d 1D powder patterns after azimuthal averaging of diffraction images (c) for the different temperature measured from 300 K to 100 K (upon cooling). e In yellow: temperature evolution of the magnetisation – temperature product MT as extracted from Superconducting Quantum Interface Device (SQUID) measurements; in grey: evolution of the HS fraction as extracted from the powder pattern refinement. Grey solid line is a guide for the eyes. f Temperature evolution of the mean unit cell volume as extracted from the powder pattern refinement. b, e and f: The vertical black line indicates transition temperature Tdown.
We performed XRD measurements on a single crystal (see Methods and Supplementary Note 2) between 100 K and 250 K, in order to quantify the volume jump (volume discontinuity at the first-order phase transition). The measured temperature evolution of the unit cell volume is displayed in Fig. 1b. It shows a significant volume jump of 1.6% (22 Å3 per unit cell) at the phase transition, taking place between the two thermal hysteresis limits Tdown = 163 K and Tup = 166 K, measured respectively upon cooling and upon heating (consistent with the values reported in the previous studies20). Below and above this discontinuity, the thermal expansion is also significant in both LS and HS phases. The same measurement allowed for determination of the thermal dependence of all six-unit cell parameters for each triclinic phase. Their extrapolated evolution can be described with a linear function of temperature T for both phases (Supplementary Figure 2 and Supplementary Table 1). The volume expansion coefficients for HS and LS phases were found to be respectively 0.31 Å3/K and 0.16 Å3/K (Fig. 1b). It is a considerable thermal expansion, leading to volume increase of 33 Å3 between 100 and 250 K, in comparison with 22 Å3 jump originating from the transition.
Powder XRD study at thermal equilibrium of micro-crystals
In the following, we will discuss measurements performed on micro-crystals of [FeIII(3-MeO-SalEen)2]PF6 embedded in a polymer thin film (see Methods). The small crystallites are plate-shaped, with average dimensions 3.5 µm × 0.35 µm × 0.13 µm21. The size is very much dependent on the synthesis conditions. Smaller crystallites are possible to obtain, but these were chosen to ensure diffraction patterns of sufficient quality for a quantitative analysis. The crystallites were dispersed in polyvinylpyrrolidone (PVP) polymer matrix and the composite films were spin-coated on a glass substrate, as described in26.
The powder XRD measurements were performed at ESRF, ID09 beamline. XRD images were recorded on a 2D detector in quasi-grazing reflection geometry at 0.2° incidence angle (see Methods). This experimental geometry was used to reduce the diffuse background due to scattering from the glass substrate and thereby enhance the diffraction from the thin film sample. A typical diffraction image is shown in Fig. 1c. The diffraction rings from the polycrystalline sample are clearly visible, with a patterning due to preferred orientation of the micro-crystals. Indeed, since the micro-crystals are plate-shaped, they tend to align with the shortest dimension (crystallographic c axis) perpendicular to the film surface. Several micro-crystals can also stack in-depth.
Temperature-dependent steady states measurements were first performed to characterize the XRD signatures of the phase transition in the polycrystalline sample. The measured diffraction patterns are shown in Fig. 1d. Strong diffuse background, mainly attributed to PVP (Fig. 1c) was removed, and the important peak broadening was ascribed mainly to a large X-ray footprint at the small angle of incidence. The latter made the analysis challenging, yet as detailed below, the full-pattern refinement was possible. This allowed for retrieval of key parameters, namely the phase fraction and the volume change.
To this end, we applied the Pawley approach27 using the Topas software28, and a similar method to the one recently applied for powder diffraction studies of photo-induced structural changes29,30. The refinements details are provided in Methods. In short, this method constrains the Bragg peaks positions according to the refined unit cell parameters, but allows the Bragg peaks intensities to vary freely. At room temperature, the sample is fully in the HS phase. At 100 K, it was not possible to fit the pattern satisfactorily considering LS phase only, therefore a biphasic state had to be considered, in contrast with the bulk single crystal. We ensured the stability of the refinement further by parametrization of the unit cell and peak intensities, as detailed hereafter. Firstly, to describe the volume evolution of both phases as a function of temperature T, the respective six unit cell parameters were forced to follow the thermal expansion determined with the single-crystal study and shown in Supplementary Figure 2. Consequently, the temperature T was the only parameter required to describe the evolution of all unit cell parameters. Secondly, the relative Bragg peak intensities Ihkl were fixed for both phases: Ihkl,HS, and Ihkl,LS were estimated from the refinements of the patterns at room temperature and 100 K, respectively. Thirdly, the complete powder patterns of a biphasic state (LS, HS) were fitted with the high spin fraction XHS as the only refined parameter (see Methods). In such a case, XHS accounts both for intensity changes and peak shifts, since the thermal expansion is phase-dependent.
The refined XHS is plotted in Fig. 1e as a function of temperature. A clear change of slope is observed at a temperature close to Tdown as defined above for the bulk single crystal. Above Tdown, the sample is almost fully in the HS phase, while a more rapid decrease of the HS fraction occurs below. Note that the absolute temperature on the sample between different setups (SQUID, XRD, optical spectroscopy), and also transition temperatures due to different sample batches of microcrystals, can vary by a few 10 s of K. In order to allow comparison between the different techniques, temperature scales are shifted for all XHS curves to have the change of slope at Tdown. To avoid confusion these rescaled temperatures will be referred to as T*. With this reference, the observed XHS evolution correlates very well with the M.T product (M, magnetisation and T, temperature) measured by SQUID (Superconducting QUantum Interference Device, see Methods), directly probing the evolution of the fraction of molecules in the HS state. Thus the spin state conversion in these micro-crystals shows two peculiar features: a gradual conversion (compared to abrupt in bulk), and incomplete conversion at low temperature. The residual XHS at T* = 100 K is equal to (34 +/− 3) %, which is consistent with the previous reports26. These two features may originate mainly from non-homogeneous stress due to surface interaction with polymers, resulting in a broad regime of phase coexistence21,31. The refined XHS also allows to calculate the average volume:
where VHS and VLS are the unit cell volume of the HS and LS phase respectively.
Concurrence of the gradual spin-state conversion and thermal expansion leads to a very smooth evolution of Vaverage. Yet, the latter slightly changes slope around T* = 160 K (Fig. 1f).
Thus parametrized powder pattern refinement allows extracting accurate values for both XHS(T) and Vaverage(T) simultaneously, and the ensuing discussion is hinged upon it. We should nonetheless mention some underlying assumptions. First hypothesis is that the LS and HS phases can be treated as separate diffracting domains. Explicitly, XHS quantifies the fraction of ordered HS domains, rather than counting the HS molecules. In case of random distribution of HS and LS molecules in the crystals, this model would lead to a misestimation of the real molecular fractions. A good correlation between XHS and the fraction obtained with SQUID, counting individual molecules irrespective of their spatial ordering, supports our assumption. The second hypothesis is that other structural distortions upon temperature within a given phase have negligible contribution to the changes of Ihkl. This is substantiated with measurements on a single crystal (Supplementary Figures 1.1 and 1.2) and therefore seems reasonable too.
Time-resolved XRD study of photoinduced dynamics
The tr-XRD images were recorded at 100 K with the setup described above. The crystallites were excited with 1 ps pump laser at 800 nm, as in previous optical studies18. The experimental time resolution was limited by the X-ray pulse duration to 100 ps. The details about the setup and data reduction are given in Methods.
The time-resolved patterns covering several decades in time are shown in Fig. 2a. Photo-induced changes can be seen on these patterns for all positive delays, and they are emphasized in the difference patterns (Fig. 2b). The comparison with steady-state diffraction patterns measured at low and high temperature allows for a qualitative description. The shape of the difference patterns can be explained by a shift and a change of the diffracted intensity, from LS- to HS-Bragg peaks. These structural changes are clearly visible in Fig. 2b) for the intense peaks indexed (101), (01-1), and (102). In contrast, it is difficult to exploit the photoinduced change of (111) peak given the precision of our measurements. However, the peak shift to smaller q due to volume expansion would produce a similar difference pattern. Therefore, separating these two possible contributions requires more quantitative analysis as described below. A closer inspection of the difference patterns also gives some insight into the dynamics. First changes occur within the 100 ps time resolution. Thereafter, the difference patterns change shape, suggesting a sequence of processes with structurally distinct signatures.
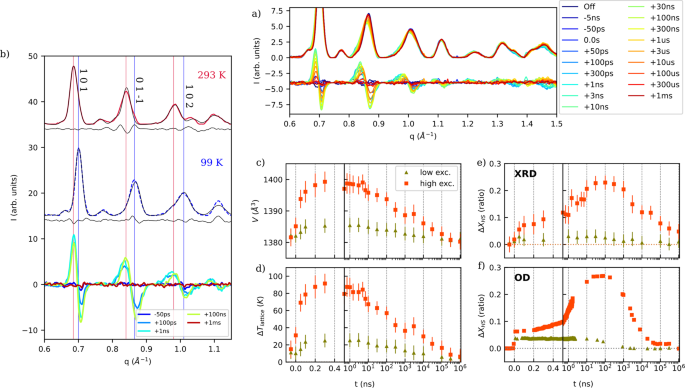
a Time-resolved powder patterns and the corresponding difference patterns measured at the rescaled temperature T* = 100 K under high excitation fluence 380 μJ/mm2 (difference patterns are multiplied by 2 and shifted by 4 for clarity). Blue to red colors correspond to increasing time delay from negative to positive (detailed delays in Methods). b Integrated steady state patterns measured at room temperature (High Spin (HS) state, in red, plain line) and T* = 100 K (in blue, dotted line), offset for clarity by arbitrary values. Black lines are the result of powder pattern refinements (see Methods) and grey lines correspond to the residual (difference between experimental and refined patterns). Time-resolved differential pattern measured at T* = 100 K from −50 ps to 1 ms after photo-excitation under high fluence 380 μJ/mm2 (multiplied by 4 for clarity), showing the characteristic shape for volume expansion (peak shifts towards lower q values). Red and blue vertical lines indicate the position of major HS and Low Spin (LS) Bragg peaks, respectively. Time evolution of the unit cell volume (c), temperature change ∆Tlattice (d, see Methods), and relative HS fraction ∆XHS (e) as extracted from powder pattern refinement. Error bars correspond to 3σ, where σ is the refinement error as defined in Topas (see methods). f time evolution of ∆XHS at T* = 110 K (see Supplementary Note 7) as extracted from optical spectroscopy (adapted from18). In orange (squares): high excitation density (380 µJ/mm² and 410 µJ/mm² for X-ray diffraction (XRD) and optical density (OD) measurements, respectively), in dark green (triangles): low excitation density (60 µJ/mm² and 100 µJ/mm² for XRD and OD measurements, respectively). For this low fluence, the ΔXHS is multiplied by 2 for clarity.
In order to analyze the underlying structural dynamics, the same method of full-pattern refinement as in the temperature study was applied to the tr-XRD patterns. The refinement results are shown in Fig. 2c. The parametrized model was similar to that used in the temperature study. However, in addition to XHS, the lattice temperature becomes an adjustable parameter Tlattice, to account for the heating which drives the non-equilibrium lattice expansion in each phase (see Methods). The time evolution of the structural parameters obtained for the highest excitation density (380 µJ/mm²) reveals multistep dynamics. A small increase of ΔXHS estimated at 7%, accompanied by a small volume increase, is observed at the early step (i.e., within 100 ps time resolution). Even if the initial volume rise cannot be accurately determined due to the 100 ps time resolution, the maximum of the volume expansion is sufficiently pronounced and well resolved at around 300 ps. At this delay, for high excitation density, the refinement yields a value of ΔTlattice = (90 +/− 5) K, corresponding to ΔVaverage = 19 Å3. During this second step, ΔXHS increases very little, even at high excitation density. At such excitation, a more pronounced increase of ΔXHS occurs during a third step on the nanosecond time scale, with a plateau between t = 20 ns and t = 300 ns, corresponding to the maximum transformation ΔXHS = 23%. Finally, ΔXHS and volume decrease simultaneously, and recovery of the values at thermal equilibrium occurs on a sub-millisecond time scale. The most striking observation is the decoupling in time between the dynamics of volume change and ΔXHS: the increase of ΔXHS occurs two orders of magnitude later than the increase of volume. Importantly, their respective dependence on the excitation density is very different. Irrespective of excitation density, either low or high, the volume expansion exhibits a maximum around 300 ps. In contrast, ΔXHS exhibits a continuous decrease in the ns range at low excitation density, and a pronounced increase to a clear maximum at 20 ns for high excitation density (Fig. 2c). However, no significant increase of average volume is observed when ΔXHS reaches its maximum.
The validity of such analysis of non-equilibrium dynamics whereby Ihkl and the lattice expansion are a-priori assumed, can be questioned. Both affect mainly the refined XHS. In particular, the newly formed HS molecules might be homogeneously distributed within the crystal, rather than organized into separate diffracting phases. This homogeneous distribution would lead to a change of the average structure factor of each phase32, that is not taken into account in our analysis. Since the refinement of the structure factor (i.e Rietveld refinement) was not feasible with our data set, we instead allowed some freedom on the intensities of the LS peaks to account for a possible switching of randomly distributed LS molecules to the HS state. The resulting volume change and ΔXHS are plotted in Supplementary Figure 3. This approach leads to higher refinement errors and different ΔXHS (15%, instead of 25% in the two-domain model) but it retains the main features of the photo-induced dynamics: the maximum of volume change occurs around 300 ps while the maximum of ΔXHS change occurs around 20 ns. Moreover, as already stressed, the evolution of ΔXHS obtained by such analysis is consistent with previous optical measurements (Fig. 2f) that yield ΔXHS directly whether or not phase separation should take place. Such agreement strengthens the validity of the global analysis.
“