

https://doi.org/10.1039/D1SC06467C
”
Solid-state NMR spectroscopy
High-resolution solid-state 13C NMR data were recorded at ambient temperature (20 °C) for samples of the α polymorph and the β polymorph of l-tyrosine on a Bruker AVANCE III spectrometer at the U. K. High-Field (850 MHz) Solid-State NMR Facility (13C Larmor frequency, 213.8 MHz; 4 mm HX probe; zirconia rotor; MAS frequency, 12 kHz) using ramped 1H → 13C cross-polarization with 1H decoupling (using SPINAL-64) applied during acquisition. The total number of scans acquired was 32 for the α polymorph and 224 for the β polymorph, with a recycle delay of 60 s between each scan. The 13C NMR data were referenced using l-alanine, for which the carboxylate resonance was set to 177.9 ppm.
”
”
Furthermore, the isotropic solid-state 13C NMR chemical shifts calculated using DFT-D/GIPAW methodology (see Methods) for the crystal structure of the β polymorph (Table S5†) are in close agreement with the isotropic chemical shifts in the experimental high-resolution solid-state 13C NMR spectrum of this material, as shown in Fig. 5. Thus, in addition to being in excellent agreement with the experimental powder XRD data, as presented above (Fig. 4), our reported crystal structure of the β polymorph of l-tyrosine is also confirmed to be in excellent agreement with the experimental solid-state 13C NMR data.
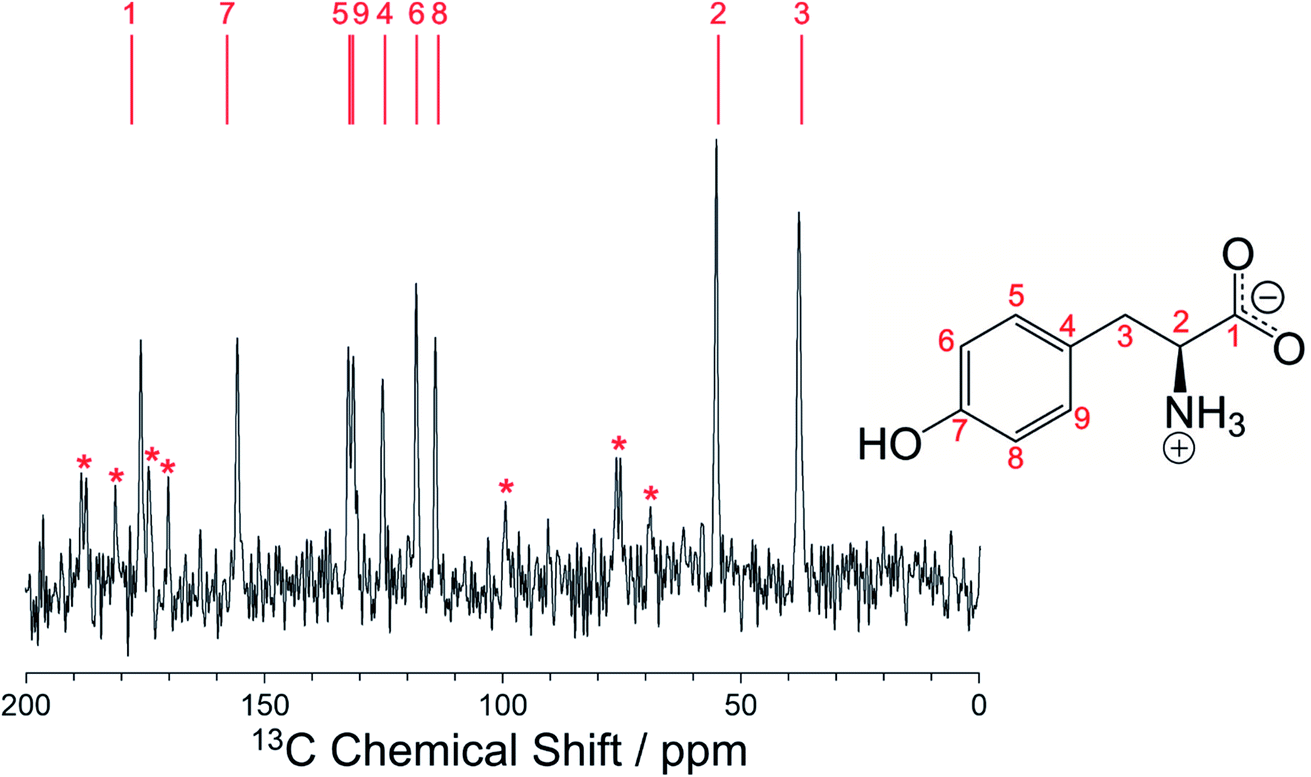
We note that the high-resolution solid-state 13C NMR spectrum of the β polymorph (Fig. 5) is clearly distinct from that of the α polymorph (Fig. S10†), and in each case there is good agreement between the experimental solid-state 13C NMR spectrum and the isotropic 13C NMR chemical shifts calculated from the crystal structure using DFT-D/GIPAW methodology (Table S5†). These observations highlight the utility of DFT-D/GIPAW calculations to compute reliable solid-state NMR data for known crystal structures, both in the context of polymorph characterization and as a powerful strategy in conjunction with structure determination from powder XRD data.
“